Untargeted LC-MS Workflow¶
The workflow proposed herein is intended as a general pipeline for untargeted LC-MS (or LC−MS/MS) data preprocessing. The main goal is essentially to turn the highly-complex LC-MS raw data into a list of features, and corresponding signal intensity, detected across the analysed samples. Such feature lists can then be exported for further downstream analysis (e.g., identification, search against spectral libraries, statistical analysis, etc.). A schematic representation of the workflow is shown below:
Tip
This workflow produces Feature lists as their result. Feature lists are a central part in visualizing the results in the Statistics dashboard and in the Interactive network visualizer.
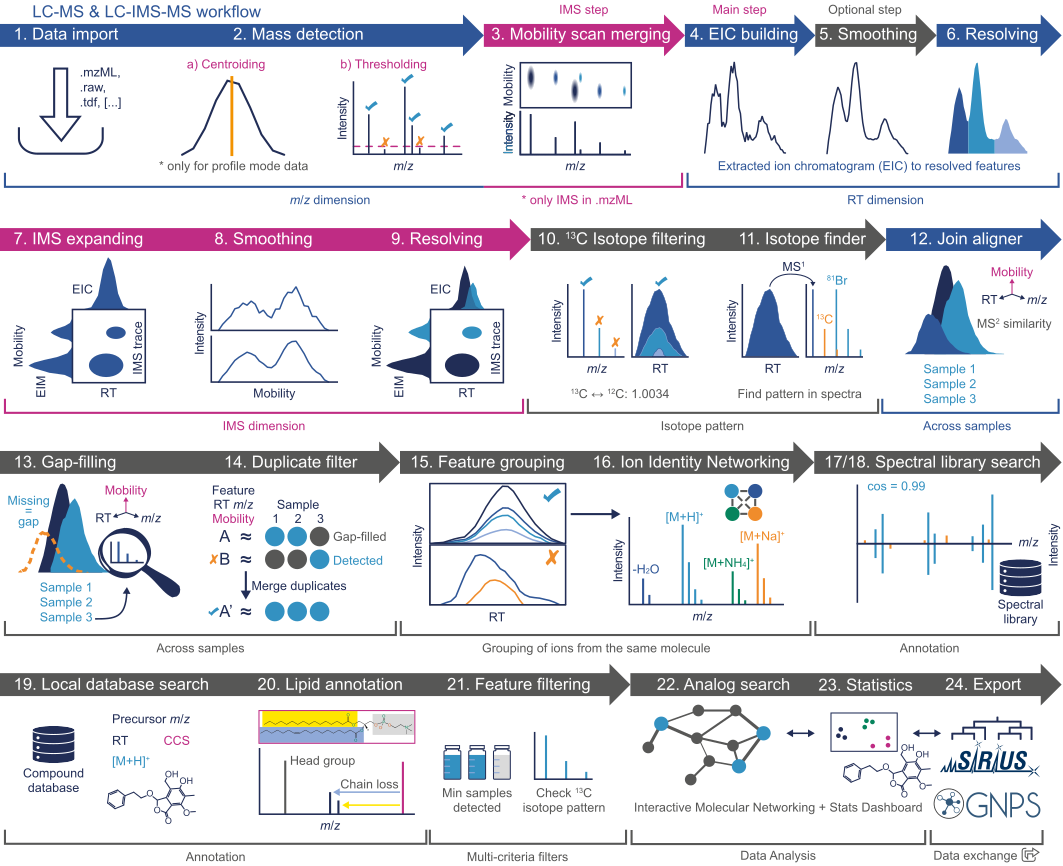 The magenta steps only refer to ion mobility data processing and are omitted here.
The magenta steps only refer to ion mobility data processing and are omitted here.
Raw data processing¶
The raw data processing consists of essentially two steps: Data import and Mass detection
Raw data import¶
Either open (e.g. mzML) and native vendor (e.g. Thermo, Bruker) data formats can be imported in mzmine. All the supported formats can be found here.
Mass detection¶
This step produces a list (referred to as "mass list") of the m/z values found in each MS scan across the LC run that exceed a user-defined threshold (i.e. noise level). For more details see the Mass detection module.
Feature processing¶
The goal of the "Feature processing" is to obtain a list of all the detected features (characterized by a RT and m/z value) from the raw LC-MS data.
Chromatogram building¶
The first step in the "Feature processing" is to build the so-called extracted ion chromatograms (EICs) for each detected mass (see "Mass detection"). For this, use the Chromatogram builder module.
The "detected" features in each file are listed in the so-called "feature lists", which are then further processed and aligned to connect corresponding features across all samples.
Smoothing in retention time dimension (optional)¶
Depending on the LC peak shape (i.e. data noisiness), the user can perform smoothing in retention time dimension. For more details see the Mass detection and Smoothing modules.
Feature resolving¶
Feature resolving step enables separation of co-eluting and overlapping chromatography peaks and as such is one of the pivotal steps in data preprocessing. For more detalis on the algorithm used and parameters settings, see the Local minimum resolver module.
13C isotope filter (isotope grouper)¶
In order to remove redundant features, such as the ones generated due to the presence of isotopologues, isotope filter should be applied. 13C isotope filter (isotope grouper) removes 13C isotope features from the feature list. Use the isotope finder for more sensitive detection of possible isotope signals.
Isotope pattern finder¶
Isotope pattern finder searches for the isotope signals of selected chemical elements in the mass list of each feature. The isotope pattern detected by the isotope finder module has priority over the one detected by the isotope filter (grouper) module, if both are available. For more details, see the Isotope pattern finder module.
Feature alignment¶
Feature alignment enables alignment of corresponding features across all samples.
Join aligner¶
This module aligns detected peaks in different samples through a match score. The score is calculated based on the mass and retention time of each peak and ranges of tolerance stipulated in the parameter setup dialog. For more information, see the join aligner module.
Gap-filling¶
Absence of features in some samples can either reflect the truth - the metabolite is absent in the given sample, or it can be due to data preprocessing.To account for this, gap filling is applied as the next step.
Gap-filling (peak finder)¶
Gap-filling can be performed on the aligned feature lists to cope with missing features that might be artifacts of the feature-detection process. For more details see the Gap-filling (peak finder) module.
Export¶
Depending on the downstream analyses, there are several export options which are accessible through Feature list methods → Export feature list.
For GNPS-Feature based molecular networking, see GNPS-FBMN or apply Interactive Molecular Networking directly in mzmine molecular_networking.md
References¶
Karaman, I.; Climaco Pinto, R.; Graça, G. Chapter Eight - Metabolomics Data Preprocessing: From Raw Data to Features for Statistical Analysis. In Comprehensive Analytical Chemistry; Jaumot, J., Bedia, C., Tauler, R., Eds.; Elsevier, 2018; Vol. 82, pp 197–225.
Pluskal, T.; Korf, A.; Smirnov, A.; Schmid, R.; Fallon, T. R.; Du, X.; Weng, J.-K. CHAPTER 7:Metabolomics Data Analysis Using MZmine. In Processing Metabolomics and Proteomics Data with Open Software; 2020; pp 232–254.
Du, X.; Smirnov, A.; Pluskal, T.; Jia, W.; Sumner, S. Metabolomics Data Preprocessing Using ADAP and MZmine 2. In Computational Methods and Data Analysis for Metabolomics; Li, S., Ed.; Springer US: New York, NY, 2020; pp 25–48.
Page Contributors¶