Spectral / Molecular Networking¶
Feature list methods → Feature grouping → Spectral / Molecular Networking
Info
This module runs molecular networking in mzmine. There is no data uploaded to GNPS in this process. For GNPS interoperability, refer to the dedicated export module export module.
This module integrates multiple popular molecular networking algorithms on MS1 and MS2 spectral relationships like modified cosine, ion identity networking, and machine learning-based models including DreaMS and MS2Deepscore2.
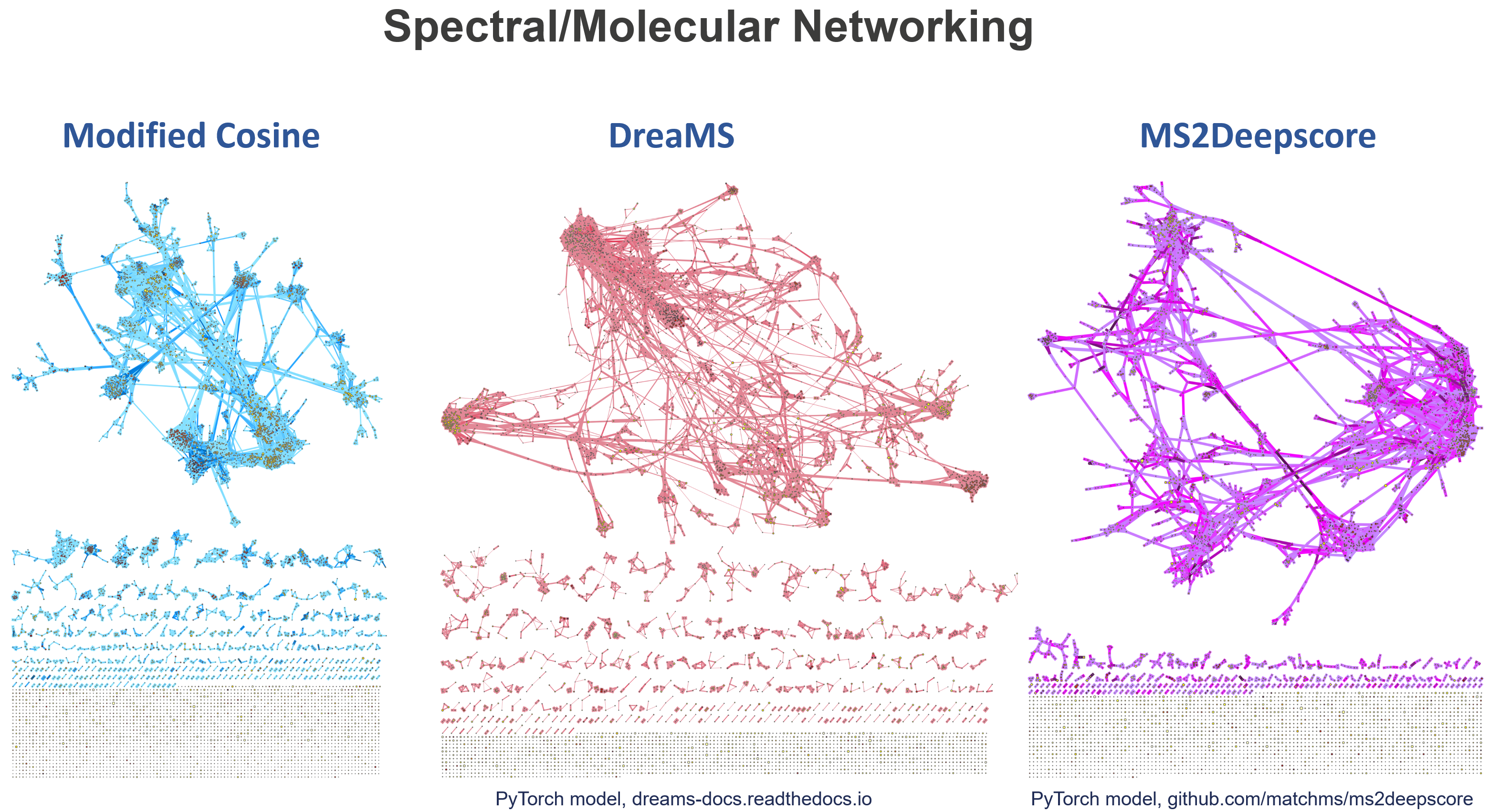
Feature-Based Molecular Networking (FBMN) and Ion Identity Molecular Networking (IIMN) group features based on their fragmentation pattern similarity. While similar to spectral library search, the algorithms used for molecular networking are open for modifications of the precursor m/zs. Choose between the modified cosine similarity that is known to connect fragmentation spectra of analog molecular structures, mostly with single modifications, or the MS2Deepscore similarity that aims to converge spectral and structural similarity.
Molecular networking can also be applied to GC/EI-MS data. Cosine similarity is used to group features, based on pseudo MS1 spectra, which can be generated by the GC-EI Spectral deconvolution module.
Warning
MS2 scans (LC-MS2) or pseudo spectra (GC/EI-MS) are required for molecular networking. For LC-MS2 make sure to run mass detection on MS2 level. For GC/EI-MS, GC-EI spectral deconvolution is required.
Finally, molecular networks can be visualized in mzmine's interactive network visualizer. Exports are available to .graphml format and to edges files (.csv).
Example of the molecular networking results visualized in mzmine.
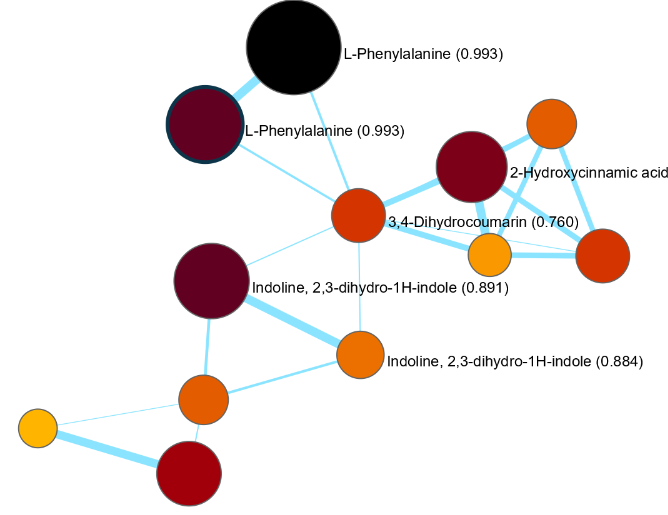
And the interactive visualizer offers more controls to style, explore, and filter networks:
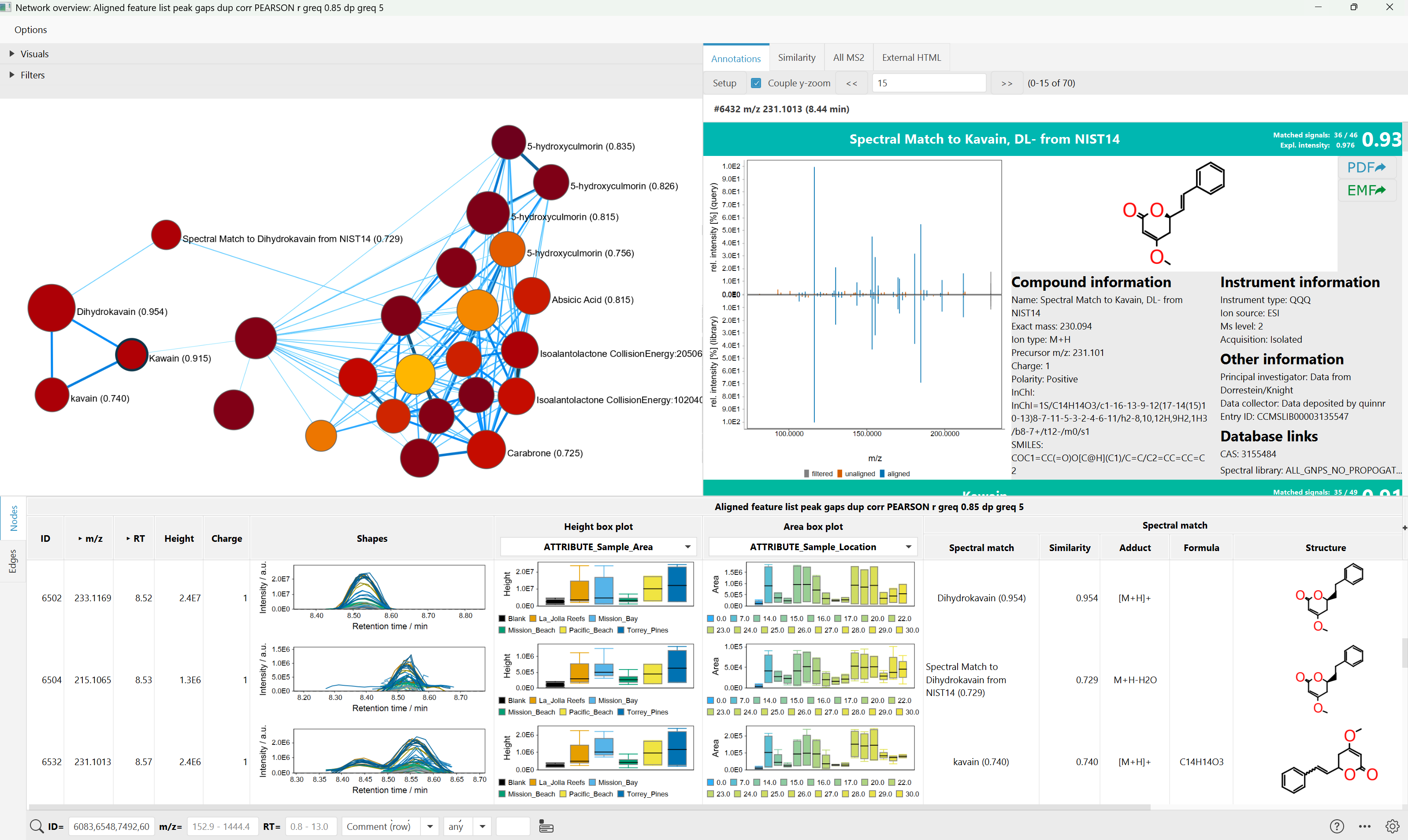
Recommended citations¶
Info
When applying Feature-Based Molecular Networking (FBMN), please consider citing:
Nothias LF, Petras D, Schmid R et al. Nat Meth 17, 905–908 (2020) https://www.nature.com/articles/s41592-020-0933-6
When applying Ion Identity Molecular Networking (IIMN), please consider citing:
Schmid R, Petras D, Nothias LF et al. Nat Comm 12, 3832 (2021) https://www.nature.com/articles/s41467-021-23953-9
When using MS2Deepscore, please consider citing:
de Jonge N, Joas D, Truong LJ, van der Hooft JJJ, Huber F bioRxiv 2024.03.25.586580
Parameters¶
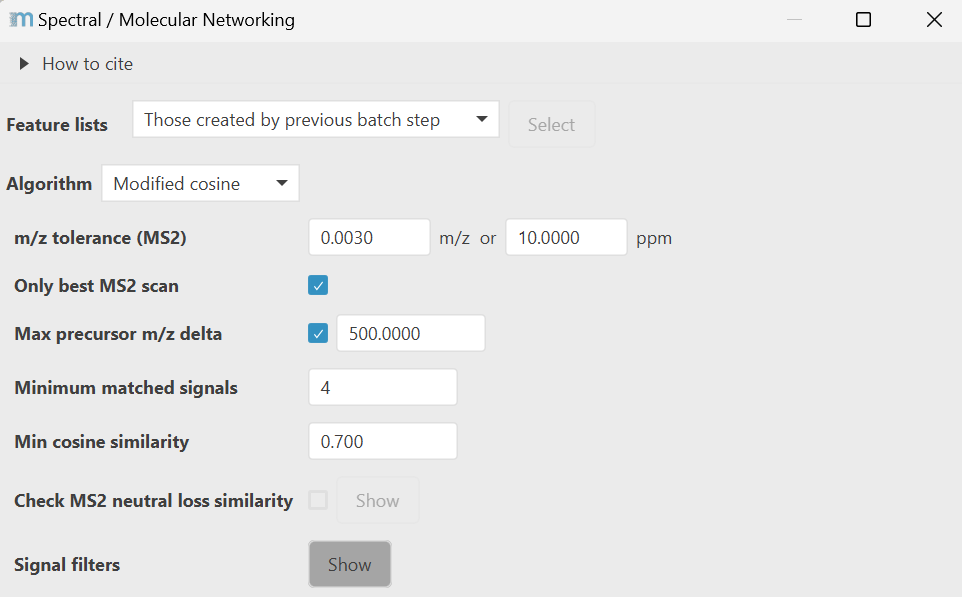
The parameter dialog changes the parameters based on the selected algorithm. Here shown for the modified cosine algorithm.
Algorithm: (Modified) cosine¶
The modified cosine similarity aligns spectra by grouping signals within the same m/z tolerance. Compared to spectral library matching, which allows only direct matches of the same fragment signals - modified cosine also considers matching neutral loss mass differences to the precursor ions. Therefore, signal pairs may be derived from the same neutral loss or the same produced fragment ion, allowing for structural analogs to cluster in the same sub networks. Signals without matching partner will be scored against 0 intensity. All spectra are square root transformed to balance the contribution of medium and high intensity signals.
m/z tolerance¶
Tolerance to group fragment signals by m/z. Fragments are group by direct match of m/z or by having the same neutral loss mass difference to the precursor ion. Most intense fragment signals are considered first.
Merge & select fragment scans¶
This parameter controls how fragment spectra are filtered, merged, and selected for downstream analysis (see detailed description). Briefly, either choose preset based spectral merging, input scans without merging, or an advanced setup for more options.
Molecular networking checks all features against each other - this is an expensive task and therefore, mzmine recommends using the Single scan: Merged across energies preset. For more density and checks, consider using the preset Representative scans which also adds scans for each fragmentation energy. The more scans the longer the task.
Max precursor m/z delta¶
If checked only features with precursor m/z difference lower than the specified value are compared.
Minimum matched signals¶
Minimum required signals to match two fragmentation spectra. Default 4 signals for small molecules. Use higher number of signals for more confident matches and compound classes with richer fragmentation data.
Min cosine similarity¶
Minimum cosine for matches between fragmentation spectra.
Signal filters:¶
Additional signal filters that reduce the spectral complexity while retaining most of the spectral information. This speeds up the matching for spectra with many fragment signals.
Remove residual precursor m/z¶
Remove all signals +- the precursor m/z.
Crop to top N signals¶
First applies the intensity filter (see below) then crops the remaining signals to the top 250.
Signal threshold (intensity filter)¶
Above N signals, start using the intensity percentile filter (see below). This does not filter to 50 signals so the final number of signals may be higher.
Intensity filter at >N signals¶
Remove signals so that X% of intensity is retained. This removes lower abundant signals that play a minor role in matching. Overall this can speed up matching.
Algorithm: MS2Deepscore¶
This algorithm uses a deep neural network to predict embeddings that aim for spectral similarities that approach structural similarities. The module allows loading of pre-trained models in the PyTorch script format. More information about MS2Deepscore can be found on github. If you use MS2Deepscore, please cite de Jonge et al. 2024 and Huber et al. 2021.
MS2Deepscore model¶
The latest model can be downloaded by clicking select -> download:
Alternatively the pre-trained MS2Deepscore model can be downloaded from https://zenodo.org/records/12628369 and contains the PyTorch script model (.pt) which should be selected in this parameter. The same folder should contain the settings.json file also supplied on Zenodo. If you want to convert a different MS2Deepscore model to a java/mzmine compatible format, you can find the relevant scripts in this repository.
Minimum signals¶
Minimum number of signals (fragments) to consider a fragment spectrum. This is not the number of matched signals because MS2Deepscore works by comparing its embeddings.
Min similarity¶
Minimum MS2Deepscore spectral similarity to consider matched. The score is scaled from 0-1 with 1 being the best match.