Feature list rows filter¶
Description¶
Feature list methods → Feature list filtering → Feature list rows filter
This module provides filters for many feature list row properties. The final resulting feature list contains rows that match all criteria or those that match none, depending on the Keep or remove rows parameter.
A range of different requirements can be set, such as the minimum number of features in the row, the minimum number of features in an isotope pattern, peak duration etc.
Feature properties are referred to the representative row values for aligned feature list with mutliple samples. This means that values like the feature's retention time are averages. Other filters may use statistics across all samples.
Parameters¶
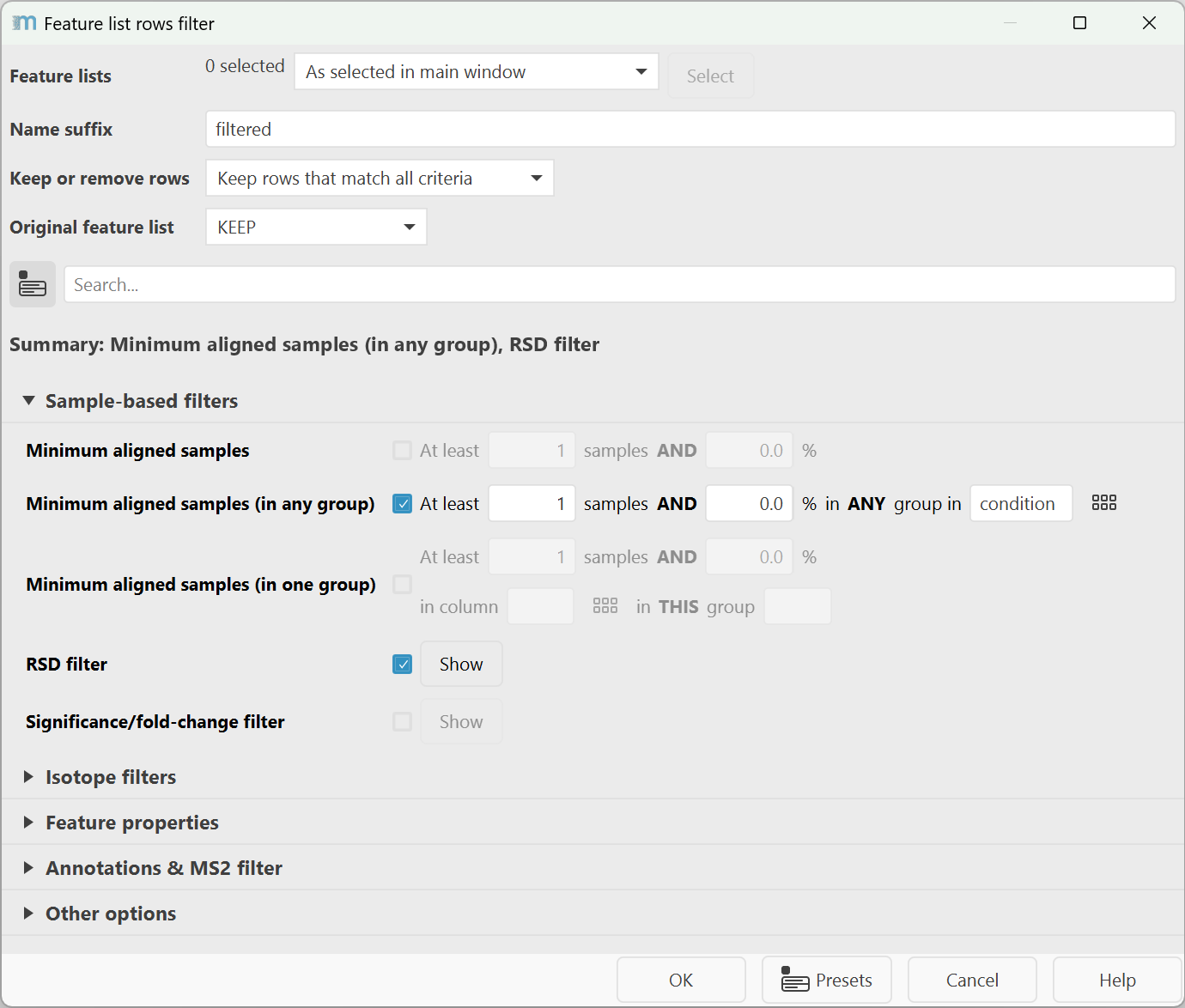
The dialog holds the most important general parameters on the top to define the behavior of these filters. All other parameters are grouped by topic and can be searched by the search bar. Selected parameters are then displayed in the summary as links that, on mouse click, provide a quick filter to find the parameters for setup.
Feature lists¶
Select the feature lists to apply the filters to.
Name suffix¶
Suffix to be added to the resulting feature list name.
Keep or remove rows¶
Select to either keep or remove the rows that match all defined criteria. Default is to keep rows that match criteria.
Original feature list¶
It can be either processed in place, kept, or removed.
Sample-based filters¶
Minimum aligned samples (previously Minimum features in a row)¶
Minimum number of samples a feature needs to be detected in a row across all samples to match. The value can be set both as an absolute number and a percentage of samples that both need to match. The percentage can help to scale the filter with increasing study sizes.
Minimum aligned samples (in any group)¶
Minimum number of samples a feature needs to be detected in any group in a metadata column. The percentage is applied for each group separately. The value can be set both as an absolute number and a percentage of samples that both need to match.
Minimum aligned samples (in one group)¶
Minimum number of samples a feature needs to be detected in one specific group in a metadata column. The value can be set both as an absolute number and a percentage of samples that both need to match.
RSD filter¶
Filters by the relative standard deviation of the abundance (height or area) in one metadata group. This filter is often used with pooled QCs.
Define a sample group by selecting a metadata column and a group in that metadata column that may only show a maximum relative standard deviation (= coefficient of variation). Only rows that show a CV below the given value, will be retained. A good sample group for this application are pooled QC samples. If a feature is not detected in all QC samples, it will be removed, unless the "Keep undetected" option is enabled.
Significance/fold-change filter ("Volcano plot filter")¶
Filter that works similar to the volcano plot on both the significance and fold-change between samples from two groups. Requires internal setup of the abundance measure, missing value imputation, significance test, and thresholds for the p-value and log2(fold-change).
Groups are selected from the project metadata, two groups in the same column.
Isotope filters¶
Isotope filters require that the 13C isotope filter and/or isotope finder modules were applied.
Minimum features in an isotope pattern¶
The required minimum number of signals in a row's isotope pattern. This is a basic filter and stems from mzmine 2 that only considers the number of signals. It is better to use the more sophisticated Validate 13C isotope pattern filter.
Validate 13C isotope pattern¶
Searches for a +1 13C signal (considering possible charge states) within an estimated range of carbon atoms. Optionally: Detect and filter rows that are 13C isotopes by searching for the preceding-1 signal.
Remove redundant isotope rows¶
Removes rows that are not the most intense or the monoisotopic peak in an isotope pattern.
Feature properties¶
m/z¶
Range of acceptable (average) m/z values in a row across all samples.
Retention time¶
Range of acceptable (average) retention times in minutes.
Chromatographic width (previously Features duration range)¶
Range of acceptable (average) feature durations as chromatographic widths in minutes.
Chromatographic FWHM¶
Range of acceptable chromatographic full-width at half maximum (FWHM) in minutes.
Charge¶
Range of row Charge
 Please, run isotope finder to determine
charge.
Please, run isotope finder to determine
charge.
Mass defect¶
Mass defect as a feature filter can be used for selective detection of compounds of interest, and the values accepted are 0.314-0.5 or 0.90-0.15.
Kendrick mass defect¶
Filter features in a Kendrick mass defect (KMD) range. For more details see Kendrick mass defect.
If KMD is used, the following parameters can be changed in the setup.
- Kendrick mass defect Permissible range of a Kendrick mass defect per row
- Kendrick mass base Enter a sum formula for a Kendrick mass base, e.g. "CH2"
- Shift Enter a shift for shift dependent KMD filtering
- Charge Enter a charge for charge-dependent KMD filtering
- Divisor Enter a divisor for fractional base unit dependent KMD filtering
- Use Remainder of Kendrick mass Use Remainder of Kendrick mass (RKM) instead of Kendrick mass defect (KMD)
Annotations & MS2 filter¶
Never remove rows with MS2¶
If checked, all rows with MS2 are retained without applying any further filters on them.
Require MS2 scan (previously Feature with MS2 scan)¶
If checked, only features that have MS2 scan will be kept.
Never remove annotated rows¶
If checked, keep all rows with annotation and skip further filtering for them.
Flexible filter (e.g., annotation)¶
This filter is defined by a search type, matching mode, and textual query. In general, annotation search types will match against any annotation like spectral library match, lipid match, or other feature annotations from other modules. All annotations are considered, this means that sometimes a row will match the filter, but the reason is not the primary annotation visible but a secondary one that may be hidden in the list of, e.g., library matches. The comment (row) search with modes * any and all* are useful options for row tagging and searching. Just insert comments in the feature table comment column and use this filter.
Only identified?¶
If the checkbox is selected, only identified compounds will be retained.
Text in identity¶
Only rows that contain this text in their annotations will be retained. Check out the flexible filter for better filtering options.
Text in comment¶
Only rows that contain this text in their comment field will be retained. Check out the flexible filter for better filtering options like substring matching against the comment (row) type.
Other options¶
Require other detector correlation¶
If checked, the rows that do not have at least one feature that is correlated to a signal of another detector will be removed.
Reset the feature number ID¶
If checked, row IDs will be reset. This can be helpful for downstream tools that require consecutive row IDs. Otherwise, the original IDs may provide better traceability throughout the mzmine data processing.